
工具二:auto_prolif.py (ProLIF 蛋白-配体相互作用指纹分析)
其实工具二是我最开始搭建的工具,用于分析蛋白质与配体之间的相互作用
1. 工具简介
这个脚本基于 MDAnalysis 和 ProLIF 库,用来批量提取 MD 轨迹中蛋白与配体之间的动态非共价相互作用(如氢键、盐桥、π-π 堆叠、疏水作用等)。
它的核心作用是帮你把枯燥的轨迹转化为直观的图表,并解决了 GROMACS 序号错位的痛点:
- 自动映射真实残基序号:GROMACS 生成
.tpr拓扑时会把残基序号打乱重排。脚本会读取你提供的一帧.pdb结构,自动将内部错位的序号完美映射回晶体学真实的残基编号,保证出图时的标签绝对准确。 - 数据提纯与过滤:程序会计算整个 MD 过程中各相互作用的发生频率(占据率),并根据你在
config.yaml中设置的阈值(如0.15代表 15%)自动过滤掉低频噪音,只保留贡献最大的 Top 10 关键氨基酸。 - 双轨输出:每个体系跑完后,会直接生成一张带有标准配色、可用于文章的高清堆叠柱状图(
.png),同时导出一份清洗好的干净数据表(.csv),方便你随时拖入 Origin 软件进行二次作图。
2. 目录结构与依赖文件
脚本运行时会读取 config.yaml 里的 base_dir_win 目录,并在当前脚本所在的文件夹下自动创建同名靶点目录来存放结果。
原始数据请确保按照以下结构存放:
E:\GROMACS\Two Target\ <-- 你的工程根目录 (config.yaml 中的 base_dir_win)
├── STAT3\ <-- 靶点名称 (config 中的 targets)
│ ├── Hit1\ <-- 具体的复合物体系 (以 config 中的 hit_prefix 开头)
│ │ ├── md_center.xtc <-- 【必须】已去除 PBC 并居中的轨迹文件
│ │ ├── md.tpr <-- 【必须】MD 运行输入拓扑文件
│ │ └── frame_0.pdb <-- 【必须】任意一帧的 PDB 文件 (脚本靠它来获取真实的残基序号)
│ └── Hit2\
└── 当前运行目录\ <-- 【运行后自动生成】
└── STAT3\
├── Hit1\
│ ├── Hit1_Occupancy_Origin.csv <-- 清洗好的数据源
│ └── Hit1_Occupancy_Stacked.png <-- 直出的堆叠柱状图
└── Hit2\
3. 操作指南
- 核对配置:打开
config.yaml,检查prolif节点下的ligand_resname(你的小分子在拓扑里的残基名,比如MOL)和occupancy_threshold(你想要的过滤阈值,默认0.15)。 - 检查文件:确认每个待处理的 Hit 目录中都有
.tpr、.xtc以及最重要的frame_*.pdb文件。 - 一键运行:在 Windows 的终端中执行:
python auto_prolif.py
* 收取结果:终端会实时打印每个体系的处理进度(如 [Hit1] ✅ 处理成功!)。全部跑完后,直接去脚本同级目录下的对应靶点文件夹里查看图表和 CSV 数据即可。
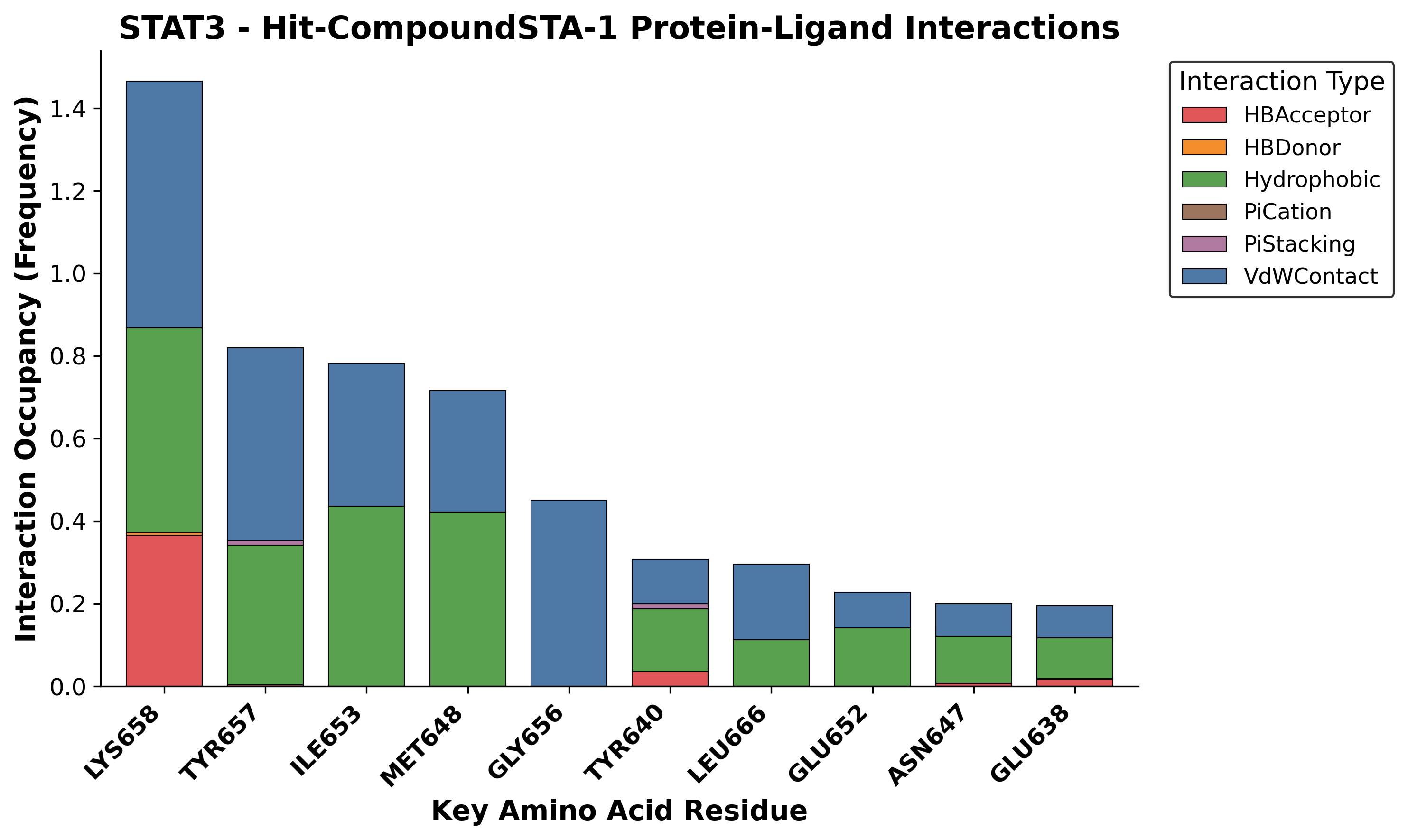
效果图如图所示
后续有考虑采用win运行下的GROMACS实现,做一个可视化程序,这样就不用直接代码运行!!